Унікальний випадок пренатальної діагностики синдрому П'єра Робена з клонічними судомами в плода

Анамнез. Дитина від першої вагітності, перших пологів. Під час вагітності: токсикоз першої половини. Обстеження на інфекції TORCH-комплексу: результати негативні. При проведенні ультразвукового дослідження в терміні 22−23 і 26−27 тижнів відзначено багатоводдя, «асиметрія у розвитку півкуль мозку плода за рахунок збільшення в розмірах лівої півкулі, розширення бокового шлуночка зліва до 16,8 мм… Читати ще >
Унікальний випадок пренатальної діагностики синдрому П'єра Робена з клонічними судомами в плода (реферат, курсова, диплом, контрольна)
ДУ «Інститут педіатрії, акушерства і гінекології НАМН України», м. Київ, Україна PERINATOLOGIYA I PEDIATRIYA.2016.1(65):68−72;doi 10.15 574/PP.2016.65.68.
У роботі наведено основні відомості про синдром П'єра Робена як рідкісну генетичну патологію з висвітленням специфічних діагностичних і клінічних аспектів. Обґрунтовано необхідність своєчасної пренатальної й постнатальної діагностики для визначення тяжкості стану плода, новонародженого та дитини з можливістю подальшого адекватного лікування. Описано два клінічні випадки: плода із синдромом П'єра Робена й пренатальними клонічними судомами, а також пацієнта 2 років із синдромом П'єра Робена й множинними структурними аномаліями центральної нервової системи. Визначено особливо небезпечні прояви патології, що призводять до летальних наслідків.
Ключові слова: синдром П'єра Робена, пренатальна діагностика, клінічна діагностика, рисунки, лікування, діти.
Уникальный случай пренатальной диагностики синдрома Пьера Робена с клоническими судорогами у плода. Клинические аспекты И. Ю. Гордиенко, Т. В. Авраменко, А. А. Шевченко, Е. Н. Тарапурова, А. А. Гребиниченко ГУ «Институт педиатрии, акушерства и гинекологии НАМН Украины», г Киев, Украина В работе приведены основные сведения о синдроме Пьера Робена как редкой генетической патологии с освещением специфических диагностических и клинических аспектов. Обоснована необходимость своевременной пренатальной и постнатальной диагностики для оценки тяжести состояния плода, новорожденного и ребёнка с вероятностью последующего адекватного лечения. Описаны два клинических случая: плода с синдромом Пьера Робена и пренатальными клоническими судорогами, а также пациента в возрасте двух лет с синдромом Пьера Робена и множественными структурными аномалиями центральной нервной системы. Определены особенно опасные проявления патологии, которые приводят к летальным исходам.
Ключевые слова: синдром Пьера Робена, пренатальная диагностика, клиническая диагностика, рисунки, лечение, дети.
The unique case of prenatal diagnostics of Pierre Robin syndrome of the fetus with clonic cramps. Clinical aspects I.Yu. Gordienko, T.V. Avramenko, A.A. Shevchenko, O.M. Tarapurova, G.O. Grebinichenko SI «Institute of Pediatrics, Obstetrics and Gynecology NAMS of Ukraine», Kyiv, Ukraine.
The paper presents basic information about Pierre Robin syndrome as rare genetic pathology with a focus on the specific diagnostic and clinical aspects. It was grounded the necessity of the timely prenatal and postnatal diagnosis for the evaluation of wellbeing of fetus, newborn and child and possibility for sub-sequent adequate treatment. There were described two clinical cases: prenatal diagnostics of Pierre Robin syndrome in the fetus with clonic cramps, and case of Pierre Robin syndrome and multiple anomalies of central nervous system in two years old patient. Especially dangerous displays are certain pathologies that result in fatal outcomes.
Key words: Pierre Robin syndrome, prenatal diagnostics, clinical diagnostics, pictures, treatment, children.
Синдром П'єра Робена являє собою симптомокомплекс, який складається з мікроретрогнатії, неповної розщілини м’якого піднебіння і глосоптозу (зміщення язика до задньої стінки глотки і вгору). При цьому розщілина піднебіння зустрічається у 90% пацієнтів із даною патологією [17]. За результатами ретроспективних досліджень, поширеність синдрому П'єра Робена становить 1 випадок на 2−30 тис. живонароджених дітей, а смертність — понад 30% випадків. У Данії, за даними літератури, рівень поширеності синдрому П'єра Робена складає 1 випадок на 14 тис. Співвідношення чоловіків до жінок дорівнює 1:1 [3, 6, 15, 24]. В Україні на сьогоднішній день немає точних даних про поширеність синдрому П'єра Робена.
Дана патологія названа на ім'я французького стоматолога П'єра Робена, який у 1923 р. на підставі власних спо-стережень визначив взаємозв'язок між мікрогнатією, розщілиною піднебіння та обструкцією дихальних шляхів. Зазначена нозологія також має назву «послідовність П'єра Робена». Це пов’язано з тим, що неповна розщілина м’якого піднебіння є вторинним дефектом унаслідок аномального розвитку (гіпоплазії) нижньої щелепи під час вагітності. Це призводить до укорочення дна ротової порожнини, язик розміщується високо в порожнині рота і запобігає повноцінному зрощенню піднебіння. Поєднання мікрогнатії і глосоптозу може спричинити утруднення дихання і труднощі під час харчування в новонародженого, унаслідок чого в багатьох випадках має місце інспіраторна обструкція дихальних шляхів [1, 9, 12, 17, 19].
Порушення ембріонального розвитку нижньої щелепи може відбуватись як унаслідок наявності механічної компресії всередині матки (рубець, фіброз, пухлина, кіста, багатоплідна вагітність), так і в разі впливу інфекції на ранніх етапах вагітності чи нейрогенетичних уражень. Слід зазначити, що вказана патологія може бути як ізольованим синдромом, так і виявленням генетичної патології. Зокрема, як частина генетичної патології синдром П'єра Робена описаний майже при 300 синдромах. Успадкування відбувається за аутосомно-домінантним і аутосомно-рецесивним типами. Якщо в батьків вже є дитина з ізольованим синдромом П'єра Робена, то вірогідність народження такої другої дитини становить 1−5%. У випадку зв’язку синдрому П'єра Робена з генетичною патологією ризик народження другої такої дитини в тих самих батьків підвищується [14, 16, 22].
Слід відзначити поєднання патології ротової порожнини при синдромі П'єра Робена з патологією інших органів і систем. Найбільш частими є аномалії слухового апарату (75% випадків) із втратою слуху в 60% хворих [5, 24].
Також описані вроджена катаракта, міопія, вади серця, сечостатевої системи, аномалії розвитку груднини та хребта, полідактилія і вроджена відсутність кінцівок [17, 24]. Аномалії центральної нервової системи мають місце в 50% випадків у пацієнтів із синдромом П'єра Робена, зокрема, затримка розвитку психіки і мовлення, рухові порушення, мікроцефалія, гідроцефалія. Частими також є епілептичні напади. Розумова відсталість має місце приблизно у 20% хворих [16, 18].
З огляду на вищенаведене, однією з найбільш актуальних проблем є своєчасна пренатальна діагностика. Слід зазначити, що візуалізація структур обличчя з виявленням аномалій розвитку стає можливою з 11−13 тижнів вагітності. У цьому зв’язку слід акцентувати увагу на тому, що мікрогнатія є важливим маркером різних генетичних синдромів із наявністю чи відсутністю хромосомних аномалій. Діагноз синдрому П'єра Робена може бути встановлений пренатально, а метод ультразвукової діагностики є інформативним щодо виявлення даної патології. Слід відзначити для пренатальної оцінки нижньої щелепи певні індекси вимірювання нижньої щелепи та кута нижньої щелепи. Також відзначено й поєд-нання гіпоплазії нижньої щелепи з багатоводдям (може бути обумовлене порушенням ковтання у плода внаслідок глосоптозу [2, 10, 11, 13, 21]. Після народження ця аномалія діагностується в перший день життя на підставі даних пренатальної ультразвукової діагностики та клінічної картини. Залежно від ступеня тяжкості синдрому може різко порушуватися дихання, що пов’язано із западанням язика, можливі часті випадки апное. У таких випадках дитина неспокійна, присутній виражений ціаноз шкірних покривів. Через порушення акту ковтання під час годування дитини може наступити задуха. Респіраторні порушення ускладнюють годування дитини. При годуванні може виникнути обструкція дихальних шляхів. Дитина намагається компенсувати обструкцію плачем і підвищенням фізичної активності, що призводить до порушення акту смоктання і збільшення енергетичних витрат. Це, своєю чергою, без лікувальної корекції спричиняє виснаження організму, поліорганну патологію і навіть смерть [2, 19, 20].
Тому в лікуванні дітей із синдромом П'єра Робена необхідно усунути обструкцію верхніх дихальних шляхів і розщілини м’якого піднебіння, відкоригувати годування. Протягом усього періоду раннього дитинства пацієнти мають регулярно спостерігатися отоларингологом, офтальмологом, неврологом, логопедом і ортодонтом. Підходи до лікування включають консервативні та хірургічні методи. Необхідним є обов’язкове укладання дітей на живіт. Також застосовується інтраназальний зонд у верхні дихальні шляхи. Хірургічне лікування складається в тимчасовій глосопексії — підтягування язика вперед із фіксацією до нижньої губи, а у вкрай тяжких випадках — і трахеостомії. Слід зазначити, що усунення розщілини м’якого піднебіння слід проводити до становлення мови, тому необхідно виконувати операції дітям у віці 6−19 місяців залежно від ступеня дихальної обструкції [4, 7, 8, 17, 23].
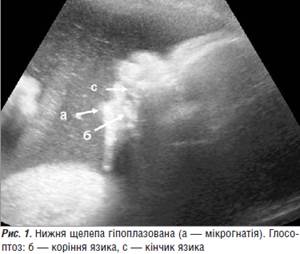
пренатальний діагностика синдром робен Клінічний випадок 1. Вагітна Б., 25 років, одноразово обстежена у відділенні медицини плода ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» в терміні 35−36 тижнів.
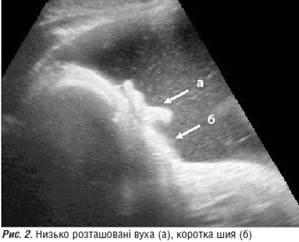
Експертне ультразвукове дослідження. Обмежена чітка візуалізація внутрішніх органів плода у зв’язку зі значним багатоводдям. Біометрія плода свідчить про відставання розмірів від терміну вагітності. Голівка плода знаходиться внизу, але під час огляду змінює локалізацію. Бокові шлуночки мозку розширені до 17 мм (вентрикуломегалія). Пренатально виявлені специфічні ознаки, наведені на рисунках 1−8: Нижня щелепа (рис. 1) гіпоплазована (мікрогнатія). Низько розташовані вуха, коротка шия (рис. 2).
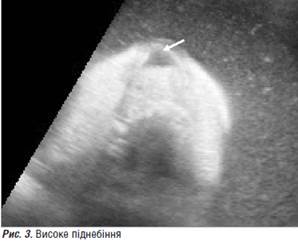
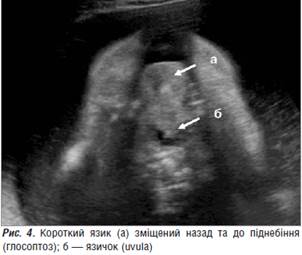
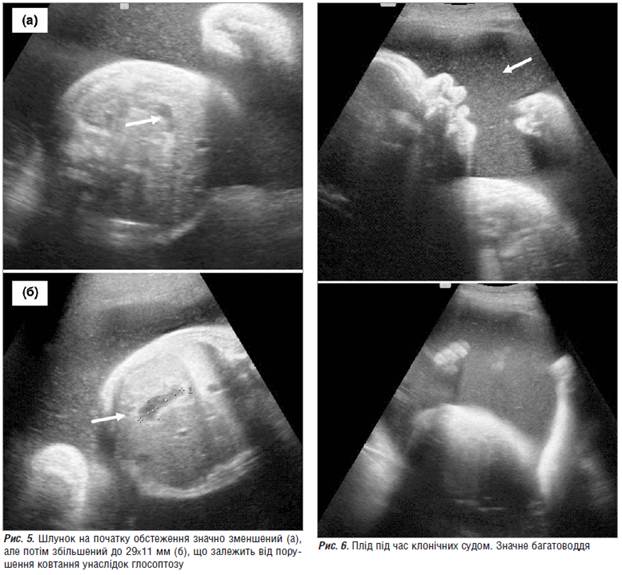
Високе піднебіння з підозрою на дефект (рис. 3). Рот відкритий, язик зміщений до задньої стінки глотки і вгору до піднебіння (глосоптоз), чергування западання та просування язика до межі верхньої щелепи/губи (рис. 4). Шлунок, незважаючи на багатоводдя, спочатку значно зменшений (рис. 5), майже не візуалізується (мікрогастрія), але потім збільшений (рис. 6) до 29×11 мм, що залежить не від патології стравоходу, а від порушення ковтання та його затримки, пов’язаної з локалізацією язика плода. Виявлені серії клонічних судом (рис. 7, 8) за участю кистей рук (пальці затиснуті в кулаки, під час огляду не розтискались). Проведений відеозапис кількох серій клонічних судом у плода. Кількість амніотичної рідини критично збільшена, у водах — велика кількість ехопозитивної суміші, амніотичний індекс (АІ) — 386 мм. Плацента розташована по передній стінці матки, ІІ-Ш ст. зрілості.
На основі даних ультразвукових досліджень встановлено консиліумом такий пренатальний діагноз: Перша вагітність 35−36 тижнів. Синдром затримки розвитку плода І ст. Синдром П'єра Робена в плода. Вентрикуломегалія. Мікрогнатія. Високе піднебіння. Порушення ковтання у плода внаслідок западання язика (глосоптоз). Пренатальні клонічні судоми. Значне багатоводдя.
Пологи проведені відповідно до акушерської ситуації.
Малюк народився з масою 2940 г, на зріст 49 см, з порушенням дихання і ковтання. Увесь час перебував на штучній вентиляції легенів, не приймав самостійно їжу, помер через 15 діб.
Клінічний випадок 2.
Пацієнт М., 2 роки, із синдромом П'єра Робена в поєднанні з множинними вродженими вадами центральної нервової системи, госпіталізований у клініку дитячої психоневрології ДУ «Інститут педіатрії, акушерства і гінекології НАМН України» з приводу вроджених вад розвитку головного мозку — гемімегаленцефалії, лісенцефалії, мозочкової лівобічної дисплазії, гіпоплазії мозолястого тіла, симптоматичної епілепсії, спастичного тетрапарезу, вираженої затримки психомоторного розвитку. Епілептичні напади — 5−6 разів на тиждень, переважно в разі засипання або прокидання. Характер нападів — асиметричні тонічні з напруженням нижніх кінцівок, витягнутою правою рукою і зігнутою лівою з подальшими клонічними посмикуваннями правих кінцівок (тривалістю до хвилини, до 12−15 разів на день). Очні яблука відведені у лівий бік.
Анамнез. Дитина від першої вагітності, перших пологів. Під час вагітності: токсикоз першої половини. Обстеження на інфекції TORCH-комплексу: результати негативні. При проведенні ультразвукового дослідження в терміні 22−23 і 26−27 тижнів відзначено багатоводдя, «асиметрія у розвитку півкуль мозку плода за рахунок збільшення в розмірах лівої півкулі, розширення бокового шлуночка зліва до 16,8 мм, ліва півкуля мозочка більша за розмірами правої півкулі, форма голівки плоду неправильної форми». У 34 тижні вагітності проведено магнітно-резонансну томографію (МРТ) малого тазу матері з висновком: МРТознаки гемімегаленцефалії плода зліва. Родорозрішення проведене в терміні 40 тижнів вагітності шляхом операції кесаревого розтину. Маса при народженні — 4100 г, довжина тіла 56 см, обвід голови — 33 см. Закричав на 3−4-й хвилині, проводилися реанімаційні заходи з подальшим перебуванням у реанімаційному відділенні протягом тижня, потім у відділенні патології новонароджених у лікарні за місцем проживання. Епілептичні напади і затримка в розвитку з народження. Серед епілептичних нападів — правобічні геміконвульсивні напади, з чотирьох місяців — асиметричні тонічні спазми, після року — генералізовані тоніко-клонічні напади і ритмічні відведення очних яблук у лівий бік. Періодично — статусний перебіг, збільшення частоти нападів на фоні інфекції та гіпертермії. Перед госпіталізацією до відділення характер нападів — у вигляді асиметричних тонічних із подальшими клонічними посмикуваннями правих кінцівок і у вигляді відведення очних яблук у лівий бік. Із протисудомних препаратів на момент госпіталізації — фенобарбітал (0,01 г тричі на добу) і депакін (35 мг/кг маси тіла на добу).?
Неврологічний статус. Обвід голови — 57 см, асиметрія обличчя, ознаки синдрому П'єра Робена (гіпоплазія нижньої щелепи глосоптоз, розщілина верхнього піднебіння, висока готичне піднебіння). За предметом не стежить. Очні щілини, S D. Парез n. oculomotorius лівого ока. Тонус у кінцівках підвищений, більше зліва. Сухожилкові рефлекси, D.
Консультація офтальмолога. Очні щілини, S D, оптичні середовища прозорі, диски зорових нервів бліді, межі чіткі, різко звужені судини. Діагноз: субатрофія зорових нервів обох очей, парез n. oculomotorius лівого ока.
Цитогенетичне дослідження (матеріал для дослідження — периферична кров): нормальний чоловічий каріотип (46, ХУ).
Електроенцефалографія (ЕЕГ). На фоновій ЕЕГ домінує високовольтна тета-активність, альфа-активність зі згладже-ними зональними відмінностями. Білатерально-синхронні спалахи високовольтної повільнохвильової активності з акцентом у лівій скронево-тім'яній ділянці. Виражена дисфункція мезенцефально-стовбурових структур.
Магнітно-резонансна томографія. На серії МР-томограм головного мозку — нерівномірне збільшення розмірів пів-куль великого мозку і мозочка ліворуч. У лобній і тім'яній ділянках лівої півкулі великого мозку — згладженість рельєфу борозен; кора нерівномірно потовщена. Мозолясте тіло в передніх відділах потовщене, в задніх — різко витончене. Гіпертрофована чотиригорбикова пластинка. У білій речовині півкуль великого мозку — підвищення інтенсивності МР сигналу на Т2 ЗЗ, обумовлене недосконалою мієлінізацією. Базальні ядра ліворуч не диференційовані. Калібр мозкових артерій зліва збільшений порівняно з правими. Нерівномірно потовщена кора лівої півкулі мозочка. МР-сигнал від білої речовини лівої півкулі мозочка на Т2 ЗЗ нерівномірно гіперінтенсивний. Диференціація — біло-сіра речовина в лівих півкулях великого мозку і мозочка порушена. Правий боковий, ІІІ і !V шлуночки деформовані. Потилична доля півкулі великого мозку і задні відділи півкулі мозочка ліворуч поширені в правий бік за середню лінію до 1,8 см. Зорові нерви, хіазма, гіпофіз, стовбурові відділи мозку — без особливостей. Розширені підпавутинні простори над півкулями великого мозку. Визначена асиметрія розмірів лицьового черепу за рахунок збільшення розмірів верхньої щелепи ліворуч, значного збільшення об'єму підшкірної жирової клітковини в лівій ділянці щоки. Розщілина верхнього піднебіння і зменшення розмірів нижньої щелепи, зміщення кореня язику в задній бік (глосоптоз).
Висновок: МРТ-ознаки поєднаної конгенітальної мальформації: гемімегаленцефалії, лісенцефалії, мозочкової дисплазії ліворуч, агенезії мозолистого тіла, аномалії розвитку лицьового черепа і м’яких тканин обличчя ліворуч, синдрому П'єра Робена.
Нижче на рис. 9 наведено кілька МР-томограм даного пацієнта, які відображають патологічні зміни (корональна, аксіальна, сагітальна проекції).
У процесі лікування (нейропротекторна, антисудомна, метаболічна терапія) проведена корекція антисудомної терапії з відміною фенобарбіталу, зменшенням дози депакіну до 300 мг на добу і призначенням топірамату за схемою шляхом поступової титрації до 5 мг/кг маси тіла на добу. Відзначене зниження частоти епілептичних нападів до 2−3 разів на місяць, дитина стала більш спокійною, поліпшився сон.

Таким чином, у даному випадку в дитини виявлено синдром П'єра Робена в поєднанні з множинними структурними аномаліями центральної нервової системи у вигляді гемімегаленцефалії, лісенцефалії, мозочкової дисплазії ліворуч, агенезії мозолистого тіла, аномалій розвитку лицьового черепа і м’яких тканин обличчя зліва. У доступних літературних джерелах немає опису синдрому П'єра Робена в поєднанні з такими множинними структурними аномаліями центральної нервової системи. Характерними особливостями даного випадку є ранній початок епілептичних нападів, їх частий поліморфний характер, геміпарез і виражена затримка психоемоційного та мовного розвитку. Проведена терапія в даному випадку дала позитивний ефект у вигляді зниження частоти епілептичних нападів. У разі виявлення мікрогнатії (пренатально або в новонародженої дитини) слід пам’ятати про синдром П'єра Робена з подальшим ретельним обстеженням для виключення інших поєднаних аномалій розвитку і наданням своєчасної медичної допомоги.
Висновки
Синдром П'єра Робена може бути як ізольованим, так і виявленням генетичних аномалій, зокрема, описаний майже при 300 синдромах. Успадкування відбувається за аутосомно-домінантним і аутосомно-рецесивним типами. Пренатальна діагностика цієї патології потребує високої кваліфікації та уваги, частіше визначається в третьому триместрі вагітності. Специфічними як пренатальними, так і постнатальними ознаками є: мікрогнатія, високе піднебіння / дефект піднебіння, зміщення язика до задньої стінки глотки і вгору до піднебіння (глосоптоз), вроджені вади центральної нервової системи, клонічні судоми. У пренатальному періоді необхідно звертати увагу на маленькі розміри шлунка при значному багатоводді та диференціювати порушення ковтання, пов’язане з локалізацією язика плода (глосоптоз), від атрезії стравоходу. Особливо небезпечними проявами в плода вважаються вроджені вади центральної нервової системи, які часто провокують появу судом, і вираженим глосоптоз, що унеможливлює нормальне дихання й ковтання в новонародженого. У тяжких випадках це призводить до летальних наслідків.
Література
- 1. Тактика ведения новорожденных с синдромом Пьера Робена / Е. В. Мельникова, М. Г Карачунский, ГВ. Тамазян [и др.] // Вопросы практической педиатрии. — 2008. — Т. 3, № 5. — С. 36—37.
- 2. Bronshtein M. Ultrasonographic diagnosis of glossoptosis in fetuses with Pierre Robin sequence in early and mid pregnancy / M. Bronshtein, S. Blazer, Y Zalel, E.Z. Zimmer // Am. J. Obstet. Gynecol. — 2005. — Vol. 193. — Р 1561—1564.
- 3. Diagnosis and treatment Pierre Robin sequence: results of a retrospective clinical study and review of the literature / A. P van den Elzen, B.A. Semmerkrot, E.M. Bongers [et al.] // Eur. J. Pediatr. — 2001. — Vol. 160. — P 47—53.
- 4. Feeding and mandibular distraction osteogenesis in children with Pierre Robin sequence: A case series of functional outcomes / P. Hong, M.K. Brake, J. P Cavanagh [et al.] // Int. J. Pediatr. Otorhinolaryngol. — 2012. — Vol. 76 (3). — Р 414—418.
- 5. Gruen PM. Anomalies of the ear in the Pierre Robin triad / PM. Gruen, A. Carranza, C.S. Karmody // Ann. Otol. Rhinol. Laryngol. — 2005. — Vol. 114 (8). — Р 605—613.
- 6. Holder-Espinasse M. Pierre Robin sequence: a series of 117 consecutive cases / M. Holder-Espinasse, V Abadie, V Cormier-Daire // J. Pediatr. — 2001. — Vol. 139. — P 588—590.
- 7. Hong P A clinical narrative review of mandibular distraction osteogenesis in neonates with Pierre Robin sequence / P Hong // Int. J. Pediatr. Otorhinolaryngol. — 2011. — Vol. 75 (8). — Р 985—991.
- 8. Kam K. Surgical versus nonsurgical interventions to relieve upper airway obstruction in children with Pierre Robin sequence / K. Kam, M. McKay, J. MacLean // Can. Respir. J. — 2015. — Vol. 22 (3). — Р 171—175.
- 9. Lidsky M.E. Resolving feeding difficulties with early airway intervention in Pierre Robin Sequence / M.E. Lidsky, T.A. Lander, J.D. Sidman // Laryngoscope. — 2008. — Vol. 118 (1). — Р 120—123.
- 10. Objective diagnosis of micrognathia in the fetus: the Jaw Index / D. Paladini, T. Morra, A. Teodoro [et al.] // Obstet. Gynecol. — 1999. — Vol. 93. — Р 382—386.
- 11. Paladini D. Fetal micrognathia: almost always anominous finding / D. Paladini // Ultrasound Obstet. Gynecol. — 2010. — Vol. 35. — P 377—384.
- 12. Pierre Robin sequence: Management of respiratory and feeding complications during the first year of life in a tertiary referral centre / M. Rathe, M. Rayyan, J. Schoenaers [et al.] // Int. J.
Pediatr. Otorhinolaryngol. — 2015. — Vol. 79 (8). -;
Р 1206—1212.
- 13. Pilu G. The prenatal diagnosis of Robin anomaly / G. Pilu, R. Romero, E.A. Reece // Am. J. Obstet. Gynecol. — 1986. — Vol. 154. — P. 630—632.
- 14. Prenatal diagnosis of Pierre-Robin sequence as part of Stickler syndrome / M. Soulier, S. Sigaudy, C. Chau, N. Philip // Prenat. Diagn. — 2002. — Vol. 22 (7). — Р. 567—568.
- 15. Printzlau A. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study / A. Printzlau, M. Andersen // Cleft Palate Craniofac J. — 2004. — Vol. 41 (1). — P 47—52.
- 16. Robin sequence: a retrospective review of 115 patients / A.K. Evans,
R. Rahbar, G. F Rogers [et al.] // Int. J. Pediatr. Otorhinolaryngol. — 2006. — Vol. 70 (6). — P 973—980.
- 17. Robin sequence: from diagnosis to development of an effective management plan / K.N. Evans, K.C. Sie, R.A. Hopper [et al.] // Рediatria. — 2011. — Vol. 127. — P 936—948.
- 18. Robin sequence: mortality, causes of death, and clinical outcomes / M.A. Costa, M.M. Tu, K. P Murage [et al.] // Plast Reconstr. Surg. — 2014. — Vol. 134 (4). — Р 738—745.
- 19. Smith M.C. Prognosis of airway obstruction and feeding difficulty in the Robin sequence / M.C. Smith, C.W. Senders // Int. J. Pediatr. Otorhinolaryngol. — 2006. — Vol. 70 (2). — P 319—324.
- 20. Teoh M. First-trimester diagnosis of micrognathia as a presentation of Pierre Robin syndrome / M. Teoh, S. Meagher // Ultrasound Obstet. Gynecol. — 2003. — Vol. 21 (6). — P 616—618.
- 21. The fetal mandible: a 2D and 3D sonographic approach to the diagnosis of retrognathia and micrognathia / D. Rotten, J.M. Levaillant, H. Martinez [et al.] // Ultrasound Obstet. Gynecol. — 2002. — Vol. 19. — Р 122—130.
- 22. The genetic basis of the Pierre Robin Sequence / L. P Jakobsen, M.A. Knudsen, J. Lespinasse [et al.] // Cleft Palate Craniofac J. — 2006. — Vol. 43 (2). — Р 155—159.
- 23. Tibesar R.J. Mandibular distraction osteogenesis to relieve Pierre Robin airway obstruction / R.J. Tibesar, D.L. Price, E.J. Moore // Am. J. Otolaryngol. — 2006. — Vol. 27 (6). — P 436—439.
- 24. Wagener S. Management of infants with Pierre Robin sequence /
S. Wagener, S.S. Rayatt, A.J. Tatman // Cleft Palate Craniofac J. — 2003. — Vol. 40 (2). — P 180—185.
Сведения об авторах
Гордиенко Ирина Юрьевна — д.мед.н., проф., зав. отделением медицини плода ГУ «ИПАГ НАМН Украины». Адрес: г. Киев, ул. П. Майбороды, 8.
Авраменко Татьяна Васильевна — д.мед.н, проф., зав. отделением метаболических нарушений и патологии плода ГУ «ИПАГ НАМН Украины». Адрес: г. Киев, ул. П. Майбороды, 8.
Шевченко Александр Анатолиевич — канд.мед.н., ст. н. сотр. отделения психоневрологии для детей с перинатальной патологией и орфанными заболеваниями ГУ «ИПАГ НАМН Украины». Адрес: г. Киев, ул. Майбороды, 8, тел. (044) 483−62−24.
Тарапурова Елена Николаевна — к.мед.н., вед. н. с. отделения медицины плода ГУ «ИПАГ НАМН Украины». Адрес: г. Киев, ул. Майбороды, 8.
Гребиниченко Анна Александровна — к.мед.н., ст. н. с. отделения медицины плода ГУ «ИПАГ НАМН Украины». Адрес: г. Киев, ул. Майбороды, 8.